
中国医疗器械(含体外诊断试剂)临床试验监管相关法规体系概述
在上篇文章中,我们对中国药物临床试验监管相关法律法规体系进行了概述,本次将对中国医疗器械(含体外诊断试剂)临床试验监管相关法规体系进行概述。
一, 首先,我们将明确医疗器械的定义:
医疗器械,是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件;其效用主要通过物理等方式获得,不是通过药理学、免疫学或者代谢的方式获得,或者虽然有这些方式参与但是只起辅助作用;其目的是:
(一)疾病的诊断、预防、监护、治疗或者缓解;
(二)损伤的诊断、监护、治疗、缓解或者功能补偿;
(三)生理结构或者生理过程的检验、替代、调节或者支持;
(四)生命的支持或者维持;
(五)妊娠控制;
(六)通过对来自人体的样本进行检查,为医疗或者诊断目的提供信息。
二, 什么是医疗器械临床试验呢?
医疗器械临床试验,是指在符合条件的医疗器械临床试验机构中,对拟申请注册的医疗器械(含体外诊断试剂)在正常使用条件下的安全性和有效性进行确认的过程。
三, 是否是所有的医疗器械,都需要进行临床试验呢?
国家对医疗器械按照风险程度实行分类管理。第一类医疗器械实行产品备案管理,第二类、第三类医疗器械实行产品注册管理。并非所有的医疗器械都要进行临床试验,第三类医疗器械临床试验对人体具有较高风险的,应当经国务院药品监督管理部门批准。国家药品监督管理局根据申请人的申请,综合分析,以决定是否同意开展临床试验。
下图将展示医疗器械分类与管理:

四, 药物临床试验与医疗器械临床试验的特点及不同:
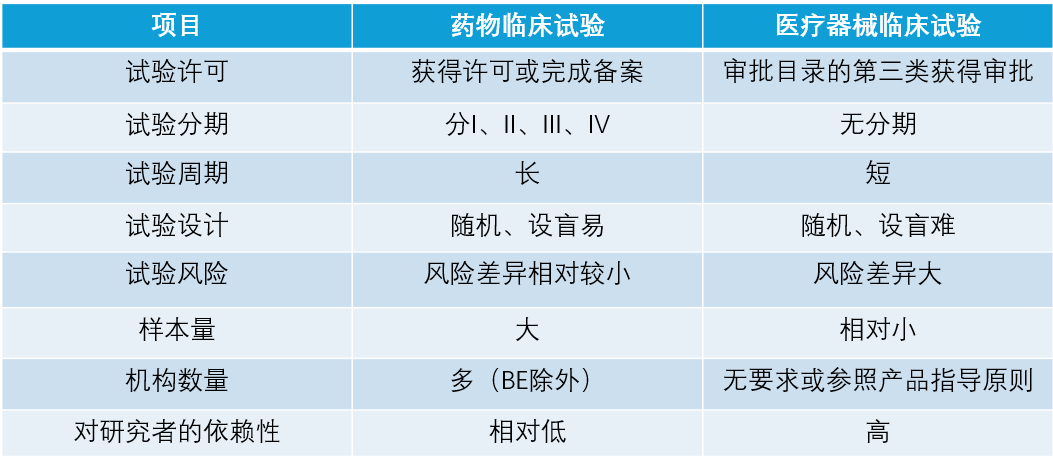
五, 在进行以上对于医疗器械的相关介绍之后,我们将重点对中国医疗器械(含体外诊断试剂)临床试验监管相关法规体系进行概述。
与药物临床试验一样,在中国医疗器械临床试验也受到一系列复杂而全面的法规体系的监管,这一体系包括不同层次的法规、规章、规范性文件和技术性指导原则。本文将按法律位阶,概述这些相关法规的名称、发布机构和常见法规,以提供对医疗器械临床试验法规体系的全面理解。
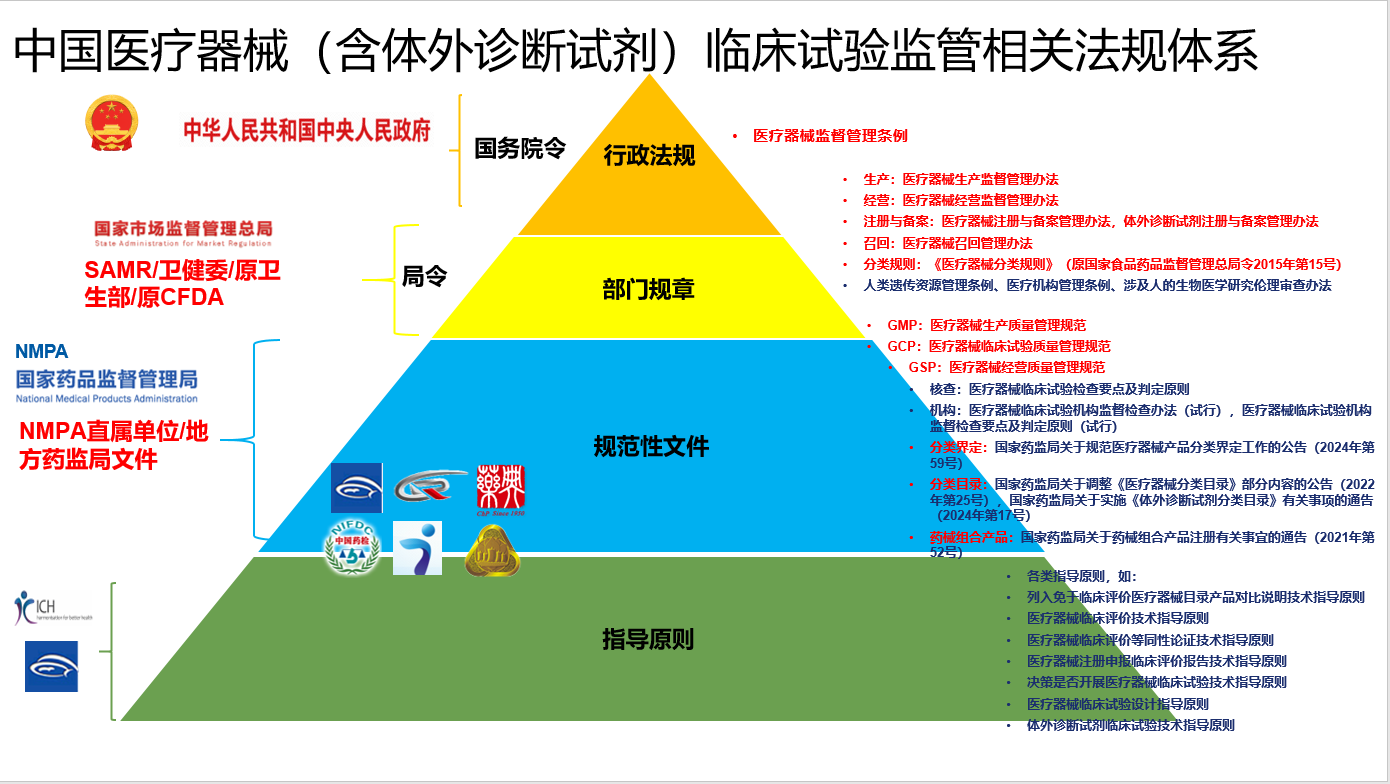
1,行政法规
2,部门规章
3,规范性文件-行政规范性文件
4,技术性指导原则
六,其他相关法律法规
涵盖了从刑事责任、民事责任、合同关系到个人信息保护的广泛内容,对于临床试验中可能涉及的各种法律问题提供了全面的法律依据。
七,结论
医疗器械临床试验法规体系不仅保护了公众健康,还促进了医疗器械产业的进步和发展。这些法规确保了临床试验的质量、效率和透明度,为医疗器械的安全性和有效性提供了坚实的法律保障。
声明:原创内容的最终解释权以及版权归晓通明达所有,欢迎转发。如需转载文章,请联系bd@hannah-associates.com。
作者简介
Fiona Yang(杨小芬)
拥有12年行业经验,其中QA经验4年。现任晓通明达质量经理,专注于GCP领域稽查。
08-09-2024



Telephone
+86 13911261194
Free consultation

Address
Room 221, 2/F Building 11, No.3 Dongbinhe Road,
Deshengmen, Xicheng District, Beijing, China
Copyright © 2020 Hannah and Associates All rights reserved
